The EU’s MDR explained for aesthetic device manufacturers
The beauty and aesthetic device market is worth billions globally, with segments like tattoo removal expected to grow by $117.09 million between now and 2027.
But devices used in surgical and non-surgical cosmetic procedures are getting a regulatory revamp in the EU due to an expansion of the EU’s enhanced Medical Device Regulation (MDR). Originally put forward in amendments to the MDR in 2022, the timeline for compliance is becoming a more pressing challenge for manufacturers who wish to continue selling in the EU.
Aesthetic devices affected by MDR
In this article, we’re not going through all the devices and treatments affected by this change. Still, as a software company, we’d like to highlight below the following devices as they often use software:
- Liposuction, lipoplasty devices.
- Light-emitting equipment, like lasers or intense pulse light (IPL) devices used in hair removal.
- Light-emitting equipment like lasers or IPL used in tattoo removal, skin resurfacing or other skin treatments.
- Brain stimulation devices.
This means that manufacturers of aesthetic devices like lasers, IPLs, and brain stimulation tools must now familiarise themselves with these requirements and ensure full compliance. This article will break down these regulations and supply a roadmap for understanding and adaptation.
Why the extra scrutiny now?
The primary goal of the MDR is to ensure that any device’s clinical benefits are greater than its potential risks. Even if beauty devices weren’t initially conceptualised for clinical uses, the EU Commission has now asked that specific aesthetic devices follow development and risk management processes, have proper labelling criteria, and proactively monitor the safety of these devices.
When MDR was initially drafted, devices like these were not under consideration, but several member states later requested changes. This was due to new available scientific evidence about the risks and side effects made possible by aesthetic treatments.
How are these devices now classified?
The above devices and treatments now fall within the following classifications:
| Device type | Rick classification |
| Liposuction, lipoplasty devices | IIb (medium high) |
| Light-emitting equipment, like lasers or intense pulse light (IPL) devices used in hair removal | IIa (medium low) |
| Light-emitting equipment like lasers or IPL used in tattoo removal, skin resurfacing or other skin treatments | IIb (medium high) |
| Brain stimulation devices | III (high risk) |
What do these classifications mean?
The classifications impact routes to gaining a CE mark and how much work is involved in meeting compliance.
Class I is for the lowest risk for devices under MDR. Having a notified body involved in making a declaration of conformity is only needed in limited circumstances. But as we can see in the table above, none of the devices being discussed here are in the lowest risk class.
Class IIa are medium-risk devices. Here, you need to get a declaration of conformity from a notified body following a conformity assessment.
Class IIb are medium to high-risk devices. The conformity assessment for these devices requires more clinical and safety related documentation to be provided to the notified body.
Class III are the highest-risk devices. These have the strictest requirements and will need a clinical trial and other technical documentation included in the conformity assessment.
What changes for manufacturers?
Aside from needing to act like medical device manufacturers, several changes must be noted:
- Comprehensive compliance: The MDR introduces a range of requirements. This includes the establishment of quality systems, collection of clinical data, creation of technical files, and initiation of post-market surveillance.
- Time is of the essence: Compliance deadlines vary based on the need for clinical trials. While some products might have a deadline stretching to June 2028, it’s worth noting that interim milestones exist from as early as 2024. Immediate preparations are needed. (We’ve got more on those deadlines further down in this article.)
- Potential Challenges: It’s essential to be aware of potential obstacles to compliance. These might include securing prompt clinical trial approvals, collaborating with notified bodies, standardising documentation processes, minimising design modifications, and understanding and integrating new responsibilities as a manufacturer.
- First steps for compliance: Before diving into the regulatory process, it’s recommended you conduct a thorough gap analysis, review your current product portfolios, then draft a well-researched action plan, and consider any necessary adjustments to business models. Here, forward planning and readying for strategic investment are critical.
- Software Standards and IEC 62304: Given the reclassification of specific beauty devices as medical devices, compliance with IEC 62304 becomes crucial. This standard pertains to the safe design and maintenance of medical device software. Adhering to this standard is seen as best practice—and the most likely to help you gain the approval of the software used in your device.
Consequences and benefits of compliance
Non-compliance carries substantial risks. These include potential patient harm, failed regulatory audits, the need for product rework, the discovery of undetected product flaws, and even the potential removal of products from the EU market.
However, compliance isn’t solely about navigating risks. It’s also about harnessing opportunities. By aligning with IEC 62304, manufacturers can enhance software practices, improve product quality, elevate safety standards, and enrich overall device usability.
An overview of IEC 62304
IEC 62304 serves as a foundational standard for medical device software, offering guidance and regulations on:
- Lifecycle processes: It gives a detailed structure for tasks throughout the software lifecycle, encompassing development, maintenance, risk management, and problem resolution.
- Risk management: Central to the standard is the identification, classification, and management of risks, categorising software based on potential harm.
- Emphasis on quality: Quality assurance processes, including verification, validation testing, defect tracking, and other related practices, are detailed to ensure software reliability.
- Documentation: A clear mandate exists for comprehensive documentation across various phases, from plans and design outlines to test cases and change logs.
- Regulatory significance: Beyond the scope of the EU MDR, the principles of IEC 62304 are recognised and revered in many jurisdictions, making it a crucial standard if you’re also selling in countries like the US.
- Adaptability: While detailed, IEC 62304 is also designed to offer adaptability, allowing it to be tailored to various software development models, including Agile.
Like MDR, IEC 62304 has its own classes. How you follow it depends on the software’s potential for harm and this will affect what class the software gains versus the overall class of the device. For IEC 62304, Class A is the lowest class and goes up to Class C (software that could cause death or severe injury).
In essence, IEC 62304 equips manufacturers with a comprehensive framework that emphasises risk management and quality, ensuring medical device software is safe and reliable. It offers a means of meeting the requirements set by the MDR when it comes to device software.
Timelines for approval
If you’re affected by these changes, and you’ve not started your journey towards compliance, you need to get started now. Deadlines for meeting MDR are fast approaching, and if your product needs clinical trials, that will add further time demands.
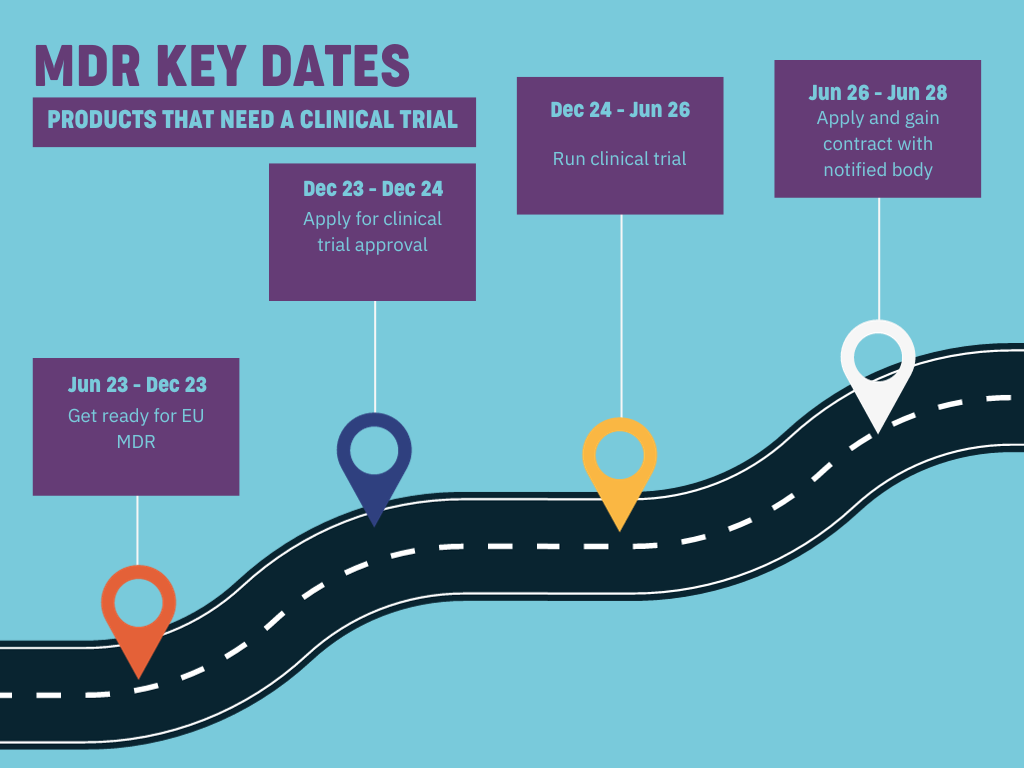
Products that need a clinical trial
Products not needing a clinical trial
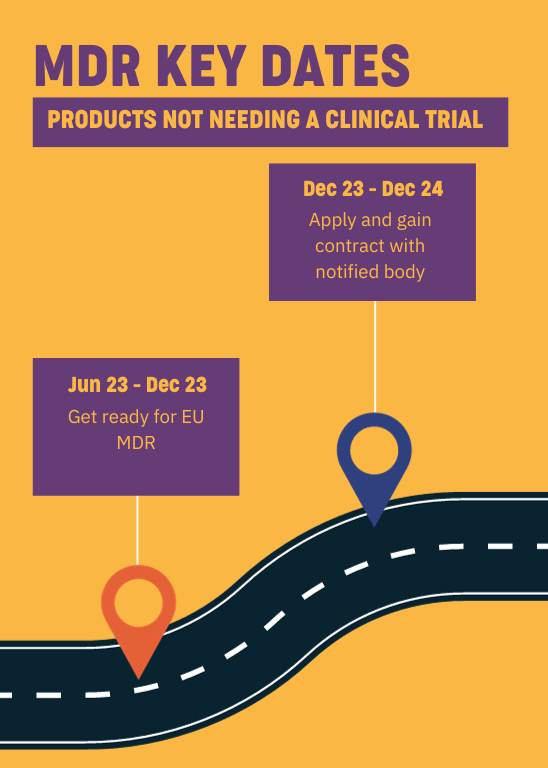
For devices not needing a clinical trial, you should be looking to prepare for MDR now. You’ll need to apply and contact a notified body from January 2024 to gain approval by June 2025.
Those with devices needing a clinical trial, like brain stimulation devices, must work to meet compliance and planning for clinical trials. That trial needs to be applied for and approved by the end of 2024 and take place before June 2026.
Considering how busy notified bodies covering the region are, and considering the upheaval that’s been caused by MDR across the sector and the backlog that was created by COVID-19 affecting the availability of notified bodies, time is on no one’s side.
Don’t let your software get in the way of MDR approval
While the road to compliance might seem challenging, with the proper understanding and proactive measures, you can not only comply but also improve the quality and safety of your products, ensuring continued success in the market.
Do you need assurances on your device software and whether it can meet what’s needed for approval? Contact Bluefruit Software today, and we can help you by performing a detailed IEC 62304 GAP analysis and set you on the path to compliance.
Alternatively, you can look at our IEC 62304 checklist to gain quick insights into whether your software hits the mark.
Find out if your product software is IEC 62304 ready
Are you wondering how well your device software holds up against IEC 62304? You can download your copy of The essential IEC 62304 checklist today.
The essential IEC 62304 checklist will help you:
- Understand critical parts of the standard from a business perspective.
- See the early signs of whether your device software is audit-ready or not.
- Discover key opportunities working to IEC 62304 presents for your business.
Success stories
Did you know that we have a monthly newsletter?
If you’d like insights into software development, Lean-Agile practices, advances in technology and more to your inbox once a month—sign up today!
Find out more




